유전자 발현량 데이터 증대를 위한 Conditional VAE 기반 생성 모델

Copyright © 2023, The Korean Institute of Broadcast and Media Engineers
This is an Open-Access article distributed under the terms of the Creative Commons BY-NC-ND (http://creativecommons.org/licenses/by-nc-nd/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited and not altered.
초록
유전자 발현 데이터는 질병의 예후 예측, 약물 반응성 예측 등 질병에 대한 이해와 정밀 의료 실현을 위한 연구들에 활용될 수 있지만 충분한 양의 데이터를 수집하는 데 많은 비용적 문제가 있다. 본 논문에서는 Conditional VAE에 기반한 유전자 발현 데이터 생성 모델을 제안하였다. 이전 연구인 WGAN-GP기반의 유전자 발현 생성 모델과 정형 데이터 생성 모델인 CTGAN, TVAE와 비교하여 본 논문의 Conditional VAE기반 모델이 생물학적, 통계학적으로 더 유의미한 합성 데이터를 생성할 수 있음을 보였다.
Abstract
Gene expression data can be utilized in various studies, including the prediction of disease prognosis. However, there are challenges associated with collecting enough data due to cost constraints. In this paper, we propose a gene expression data generation model based on Conditional Variational Autoencoder. Our results demonstrate that the proposed model generates synthetic data with superior quality compared to two other state-of-the-art models for gene expression data generation, namely the Wasserstein Generative Adversarial Network with Gradient Penalty based model and the structured data generation models CTGAN and TVAE.
Keywords:
Gene expression, Bio Big Data, Generative Model, Variational AutoencoderI. 서 론
Next Generation Sequencing (NGS) 기술의 발달로 세포 내의 유전자 발현량 (gene-expression)을 측정하기 위한 비용 감소로 인하여 수많은 개인의 유전체 분석이 가능해졌고, 다양한 환경에서 유전자 발현량 데이터가 생성되고 있다[1]. The Cancer Genome Atlas (TCGA)[2], Genotype-Tissue Expression (GTEx)[3]와 같은 프로젝트에서는 암 환자의 유전자 발현량 데이터, 정상 세포의 유전자 발현량 데이터 등 각 목표에 맞는 다양한 환자의 데이터를 축적하고 연구자들이 활용 가능 한 유전체 데이터를 공개하고 있다. 위 데이터를 이용하여 유전자 발현량의 차이 분석, 조절 관계 분석, 기계 학습 기반 예측 모델 구축 등을 통해 치주질환과 같은 질병의 예후 예측[4], 약물 반응성 예측[5], 질병 타입 구분[6], 질병과 연관된 바이오 마커 검출[7] 등의 질병에 대한 이해와 정밀 의료 실현을 위한 연구들이 진행되고 있다.
그러나 많은 양의 데이터가 축적되고 있음에도 불구하고 20,000여개의 높은 차원을 갖는 유전자 발현량 데이터를 분석하기에는 데이터의 수가 아직 부족하다. 생물학적인 시스템은 매우 복잡하고 여전히 밝혀지지 않은 생물학적 요소 간 상호작용이 많기 때문에 연구하고자 하는 조직 혹은 세포의 유전자 발현 데이터는 다양하면서도 높은 품질로 충분히 많은 양을 확보하는 것이 중요하다. 유전체 데이터는 다른 데이터들에 비해 수집 비용이 높아 수집에 어려움이 있으며 연구 범위를 특정 조직, 세포, 질병에 한정한다면 데이터의 수가 훨씬 적어져 분석이 더 어려워진다. 이를 해결하기 위한 방법 중 하나로, 생성 모델 기반의 합성 유전자 발현 데이터를 정밀하게 생성하는 것이 해결책이 될 수 있고 다음과 같은 선행 연구들이 있었다.
TCGA 데이터셋에 대한 약물 예측을 수행하기 위한 Variational Autoencoder (VAE)[8]기반의 생성 모델[9]과 합성 유전자 발현 데이터를 생성하기 위한 Wasserstein GAN with Gradient Penalty (WGAN-GP)[10]기반의 생성 모델[11]에 관한 연구가 있었다.
본 논문에서는 Conditional Variational Autoencoder(Conditional VAE)[12]에 기반한 유전자 발현 데이터 생성 모델을 제안하고 WGAN-GP기반의 모델과 합성 유전자 발현 데이터의 품질을 비교하였다. 추가로, 정형 데이터 생성 모델인 Conditional Tabular GAN (CTGAN)[13], Tabular VAE (TVAE)[13]로 생성한 합성 유전자 발현 데이터의 품질과도 비교하였다. 구체적으로, TCGA, GTEx에서 제공하는 RNA-seq 데이터셋 중 15개의 공통 조직을 선택하였다. 그 후 The Library of Integrated Network-Based Cellular Signatures (LINCS) 연구진들이 선별한 L1000 랜드마크 유전자를 선택하고 해당 데이터로 학습하여 합성 유전자 발현 데이터를 생성하고 비교하였으며, Conditional VAE기반 모델이 생물학적, 통계학적으로 더 유의미한 유전자 발현 데이터를 생성할 수 있음을 보였다.
II. 관련연구
1. Adversarial generation of gene expression
Ramon Viñas 등은 Generative Adversarial Network(GAN)[14]으로 유전자 발현 데이터를 생성하고자 WGAN-GP기반의 유전자 발현 생성 모델을 개발하고 제안하였다[11]. WGAN-GP는 Wasserstein GAN (WGAN)[15]의 문제점을 개선한 방법이다. GAN에서 생성자가 특정 값에 치우친 데이터를 생성하는 mode collapse 문제를 해결하고 손실함수가 너무 커지는 것을 막기 위한 립셔츠 제약조건(1-Lipschitz constraint function)[16]을 가중치 클리핑(Weight clipping)을 통해 적용했던 WGAN와 다르게 WGAN-GP는 경사 패널티(Gradient Penalty)를 적용한 것이 특징이다. Ramon Viñas 등은 WGAN-GP를 기반으로 하여 유전자별 유전자 발현 데이터와 각 발현에 해당하는 유전자의 조직, 암 여부 데이터를 생성자(Generator)와 비평자(Critic)가 함께 학습하여 조직과 암 여부 조건에 맞는 유전자 발현 데이터를 생성할 수 있도록 하였다.
2. Conditional Tabular GAN (CTGAN)
일반적인 정형 데이터는 연속형 변수와 범주형 변수를 함께 포함하고 있다. 정형 데이터는 수치만으로 이루어진 테이블 형식의 데이터를 의미한다. 이러한 정형 데이터를 GAN으로 생성할 경우에 연속형 변수와 범주형 변수를 동시에 생성하는 문제와 카테고리 변수에서 높은 불균형을 띄는 문제가 있다. CTGAN[13]은 Conditional GAN(CGAN)[17]을 사용해 이러한 문제점을 개선하고 합성 정형 데이터를 생성하는 GAN 기반 아키텍처이다. CTGAN의 구조는 다음과 같다.

CTGAN 모델 구조Fig. 1. Architecture of CTGAN model
CTGAN은 연속형 변수와 범주형 변수가 함께 있는 정형 데이터를 GAN으로 생성하는 문제를 해결하기 위해서 mode-specific normalization과 training-by-sampling의 방법을 제안하였다. 먼저, mode-specific normalization은 비가우시안 (Non-Gaussian) 및 다중 모드 분포(Multimodal distribution)를 따르는 연속형 변수를 전처리하기 위한 것으로, 변동 가우시안 혼합 모델(Variational Gaussian mixture Model, VGM)을 단위 분포 모델로 사용하여 각 변수의 분포 수를 추정하고 추정된 분포에 따라 변수 값을 정규화하는 방법이다. Training-by-sampling은 범주형 데이터의 불균형 문제를 해결하고 실제 데이터의 분포와 비슷한 분포로 생성하도록 조건을 부여하는 것으로, 테이블 데이터의 각 열과 범주형 변수를 [0,0,0], [1,0]와 같은 conditional vector로 원 핫 인코딩(One-hot Encoding)하여 학습하는 방법이다. CTGAN의 손실함수는 WGAN-GP와 같으며, 생성자와 판별자(Discriminator)는 conditional vector와 함께 정형 데이터를 실제 데이터와 유사하게 생성할 수 있도록 학습하게 된다.
3. Tabular VAE (TVAE)
CTGAN이 합성 정형 데이터를 생성하기 위해서 데이터를 전처리하고 학습하는 방법을 VAE에 적용한 VAE기반 아키텍처로, VAE의 손실함수를 사용한 점만 다르다.
III. 데이터셋
1. The Cancer Genome Atlas (TCGA)
미국 국립 보건원(National Institutes of Health, NIH)에서 암의 분자 생물학적 기초에 대한 이해를 증진하고자 여러 암 유형에 대한 유전체, 전사체, 단백체 데이터를 여러 기관에서 수집하여 만든 데이터베이스로 33개의 암 유형별 유전자 발현, 돌연변이 등의 데이터를 제공한다[2]. TCGA 데이터베이스의 유전자 발현량 데이터를 활용해 해당 조직의 암 데이터를 학습하고 모델링하는데 사용하였다.
2. The Genotype-Tissue Expression (GTEx) dataset
960명의 장기 기증자로부터 수집한 여러 조직의 전사체 데이터이다[3]. GTEx 데이터베이스에서 제공하는 유전자 발현량 데이터를 각 조직의 정상 데이터를 학습하고 모델링하는데 사용하였다.
3. RNA-seq
RNA-seq은 전사체를 분석하여 발현의 차이를 확인하는 방법으로, 이를 통해 RNA 염기서열을 시퀀싱하면 어떤 조직에서 어떤 유전자가 얼마나 발현되는지 확인할 수 있다.
대부분의 생물체에서 생명현상을 나타내는 서열 정보는 DNA에서 RNA로 전사(Transcription)되고 RNA가 단백질로 번역(Translation)되는 과정을 거치는데, 이 같이 DNA가 단백질 혹은 RNA을 합성하는 과정을 유전자 발현(Gene expression)이라 한다. 이러한 유전자 발현은 생명체가 처한 환경에 따라 유전자별로 늘어나거나 줄어들기 때문에 RNA-seq으로 유전자 발현량을 분석하면 어떤 생물체가 특정 환경에서 어떻게 대응하는지 파악할 수 있고 유전자의 상호작용이 어떻게 이루어지는지에 대한 연구에서 많은 도움이 될 수 있다.
4. L1000 landmark gene set
유전자 발현은 유전자에 따라 여러 세포에 걸쳐 유사하게 나타나는 경우가 있는데 이들을 분석하면 랜드마크 유전자 (Landmark gene)라 부르는 적은 양의 유전자만으로 측정없이 모든 유전자의 발현을 예측할 수 있고 선행 연구가 있다[18]. Library of Integrated Network-Based Cellular Signatures (LINCS) 프로그램 연구진은 11,350개의 목표 유전자를 가지고 있는 Connectivity Map 데이터에서 전체 유전자 발현의 약 80%를 설명할 수 있는 978개의 유전자를 랜드마크 유전자로 선별하고 L1000 landmark gene set으로 명명하였다. 본 논문에서는 L1000 landmark gene set을 사용하여 20,000여개의 유전자의 차원을 줄여 landmark gene set의 gene expression data를 합성하는 것을 목표로 하였다.
5. 데이터 통합
본 논문에서는 TCGA와 GTEx에서 제공하는 RNA-seq 데이터를 통합하고 공통 조직을 가지는 샘플을 선정하였다. 구체적으로, 두 데이터셋의 15개의 공통 조직(폐, 유방, 갑상선, 신장, 위, 간, 전립선, 타액, 식도, 식도 점막, 방광, 자궁, 자궁 경부, 췌장, S상 결장)의 데이터를 통합하여 9,146개의 샘플과 18,154개의 유전자로 구성된 유전자 발현량 데이터셋을 만들었다[19].
그 후 데이터의 수에 비해 생성하고자 하는 유전자의 차원이 너무 큰 문제가 있기 때문에 전체 유전자 발현을 잘 설명할 수 있는 landmark gene set에 한정하여 학습을 시키고 성능 비교를 진행하였다. TCGA, GTEx의 유전자와 978개의 랜드마크 유전자 중 969개의 공통 유전자를 선정하여 9,146개의 샘플과 969개의 유전자로 구성된 데이터셋을 만들었다.
6. 데이터 전처리
본 논문에서는 log2(expression_value+1), standardization을 사용해 전처리하였다. 단, CTGAN, TVAE 모델의 경우에는 같은 전처리 데이터의 학습 결과에서 -1, 1등의 정수값만 생성되는 모습을 보였기 때문에 모델의 한계점을 고려하여 CTGAN, TVAE의 입력 데이터에는 어떠한 전처리도 하지 않았다. 다만, 테스트 데이터셋을 사용한 성능 평가 시에는 CTGAN, TVAE가 생성한 합성 유전자 발현 데이터에 전처리를 적용하여 성능비교를 진행하였다.
IV. 제안하는 방법
1. Variational Autoencoder (VAE)
VAE는 변분 추론(Variational inference)을 사용하는 encoder와 decoder로 구성된 autoencoder기반의 신경망으로, 대표적인 생성 모델(Generative model) 중의 하나이다. Encoder는 입력 데이터로부터 잠재 변수 확률 분포의 모수를 추정하여 잠재 변수(latent variable)를 embedding하여 샘플링하고 decoder는 이러한 잠재 변수로부터 원래의 입력 데이터를 복원한다. 여기서 decoder는 encoder가 만든 잠재 변수의 평균과 분산을 모수로 하는 정규분포를 전제로 하여 데이터의 사후확률(posterior) p(z|x)를 학습하는데, 잠재 변수는 무수히 많을 수 있어 사후확률의 계산이 매우 어렵기 때문에 다루기 쉬운 p(z)로 근사하여 학습하는 변분 추론을 사용한다. VAE의 손실함수는 다음과 같다.
E는 기대값, p와 q는 확률 분포, DKL은 쿨백-라이블러 발산(Kullback-Leibler divergence), x는 입력 데이터, z는 잠재 변수를 나타낸다. 식 (1)의 우변의 첫 번째 항은 입력 데이터의 복원 손실(Reconstruction Error)을 의미하는데, encoder가 잠재 변수 z의 확률 분포 q∅ (z|x)를 만들면 decoder가 이로부터 입력 데이터 x를 복원하여 손실을 계산한다. 이때, 입력 데이터가 동일하더라도 매번 달라질 수 있어 발생하는 z에 대한 역전파 계산 문제를 해결하기 위해 평균이 0인 정규분포의 노이즈를 추가하여 샘플링하는 reparameterization trick을 사용하여 z을 샘플링한다. 식 (1)의 우변의 두 번째 항은 쿨백-라이블러 발산을 의미하는데, p(z|x)와 q(z) 사이의 쿨백-라이블러 발산을 계산하고 이 값이 줄어들도록 q(z)를 업데이트하여 z의 확률 분포가 정규분포에 가깝도록 근사한다.
VAE는 일반적인 autoencoder와 다르게 잠재 공간(latent space)상의 잠재 변수를 한 점 형태가 아닌 분포 형태로 나타낼 수 있기 때문에 입력 데이터를 재구성한 새로운 데이터를 만들어낼 수 있다는 장점이 있다.
2. Conditional VAE Model
Conditional VAE는 encoder와 decoder에 레이블에 해당하는 조건을 입력 데이터와 함께 넣어 학습하고 조건에 맞는 데이터를 생성할 수 있는 VAE의 변형된 모델이다. Conditional VAE의 손실함수는 다음과 같다.
레이블에 해당하는 conditional vector c가 추가된 점 이외에 나머지 수식은 VAE의 손실 함수와 동일하다.
본 논문에서는 15개의 조직 유형과 암, 정상 여부에 대한 값을 조건으로 갖는 Conditional VAE에 기반한 유전자 발현 데이터 생성 모델을 만들었다. 그림 2는 본 논문에서 만든 Conditional VAE 모델의 구조를 나타낸 그림이다.

Conditional VAE 모델 구조Fig. 2. Architecture of our Conditional VAE model
Encoder는 유전자 발현량 데이터를 조직 유형과 암, 정상 여부에 대한 정보와 함께 잠재 공간(latent space)상의 잠재변수로 embedding하고, decoder는 잠재변수로부터 새로운 유전자 발현 데이터를 재구성하여 복원하는 구조를 갖는다. 재구성된 유전자 발현 데이터는 복잡한 유전자 발현 측정값의 특성상 넓은 범위의 발현 값을 표현할 수 있어야 하므로 multivariate gaussian decoder를 사용하여 평균 제곱 오차(Mean Square Error, MSE)와 쿨백-라이블러 발산을 더한 값을 손실 함수로 사용하였다. 또한, 각 layer마다 배치 정규화(batch normalization)를 추가하여 학습이 안정될 수 있도록 하였다.
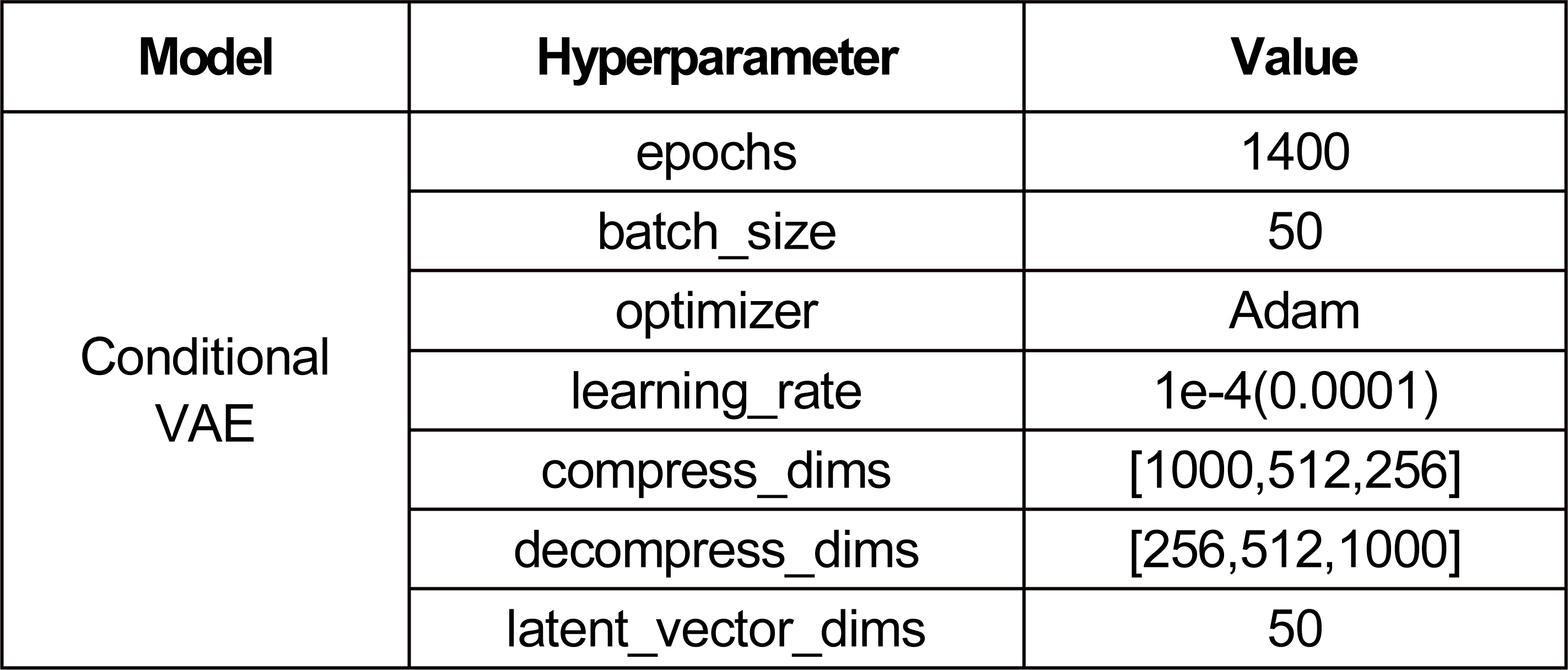
표 1은 모델에 사용한 하이퍼파라미터(Hyperparameter)에 관한 테이블이다. 에폭(epochs=1400)은 반복학습 횟수이고 배치당 예제 수인 batch_size는 50이다. Optimizer는 Adam, learning_rate는 1e-4(0.0001)으로 설정하였고 hidden layer의 수와 값에 관한 compress_dims, decompress_dims는 각각 [1000,512,256], [256,512,1000]으로 설정하였다. 잠재공간에 embedding하는 크기인 latent_vector_dims는 50으로 설정하였다.

모델에 사용한 하이퍼파라미터Table 1. Hyperparameters of our model
하이퍼파라미터는 epochs와 batch_size, latent_vector_dims는 고정하고 나머지에 대해 learning_rate는 0.001, 0.005, 0.0001, 0.00001으로, hidden layer 수는 2, 3, 4개로 나누어 학습하였고 train data를 train/ validation으로 나누어 validation data의 loss가 가장 작은 모델을 성능 평가에 사용하였다.
3. 모델 학습
본 논문에서는 전체 데이터를 7:3의 비율로 학습 데이터셋과 테스트 데이터셋으로 나누었다. WGAN-GP기반 모델, CTGAN, TVAE의 epoch는 overfitting을 고려하여 700으로 설정하였고 다른 hyperparameter는 해당 논문에서 제시한 기본 세팅으로 성능 비교 실험을 진행하였다. 학습환경은 Intel i9-10980XE CPU / 256GB RAM / Nvidia Geforce RTX 3090 / Ubuntu 20.04.5 LTS에서 진행 하였다.
4. 성능 평가 방법
각각의 모델이 생성한 합성 유전자 발현 데이터가 얼마나 잘 만들어졌는지 확인이 필요하지만 이미지와 같은 데이터처럼 사람이 직접 합성 데이터의 품질을 판단 하기가 매우 어렵다. 때문에 생물학적, 통계학적으로 더 유의미한 정보를 담고 있는지 확인하기 위한 방법으로 다음과 같은 두 가지 방법을 사용하였다.
첫째, 통계학적으로 합성 유전자 발현 데이터가 원래 유전자 발현 데이터와 유사한지 데이터의 품질을 평가하기 위해 피어슨 상관계수(Pearson’s correlation coefficient)를 기반으로 하는 gamma score로 명명한 유사도 계수(similarity coefficient)를 사용하였다[11]. Gamma score의 수식은 다음과 같다.
식3에서 A, B는 모든 유전자 사이의 쌍별 거리(Pairwise distance)를 계산한 n x n대칭 행렬이고 A와 B는 각각 합성 유전자 발현 데이터와 비교할 유전자 발현 데이터를 대상으로 한다. 식 4는 식 3에서 μ(G), σ(G)에 관한 수식이다. 식 3을 통해 A와 B의 상삼각행렬(Upper triangular matrix)에 대한 피어슨 상관계수를 계산하여 합성 유전자 발현 데이터가 원래의 유전자 발현 데이터와 얼마나 유사한지 계산하였다. 유사도는 0에서 1 사이의 값으로 표현된다.
둘째, 각각의 모델들이 조직별로 유전자 발현의 주요 특징과 암, 정상 여부의 유전자 발현량의 차이를 잘 구분하여 학습했고 각각의 모델들이 생성한 합성 유전자 발현 데이터에 제대로 표현하고 있는지 시각화하여 확인하였다. 이를 위해 차원 축소(dimensionality reduction) 알고리즘 중 하나인 UMAP[20]을 사용하여 고차원의 유전자 발현 데이터를 저차원으로 축소하여 시각화 하였다.
합성 유전자 발현 데이터를 평가할 때는 2,287개의 샘플을 가진 테스트 데이터셋과 함께 비교하였다.
V. 실험결과
1. Gamma score
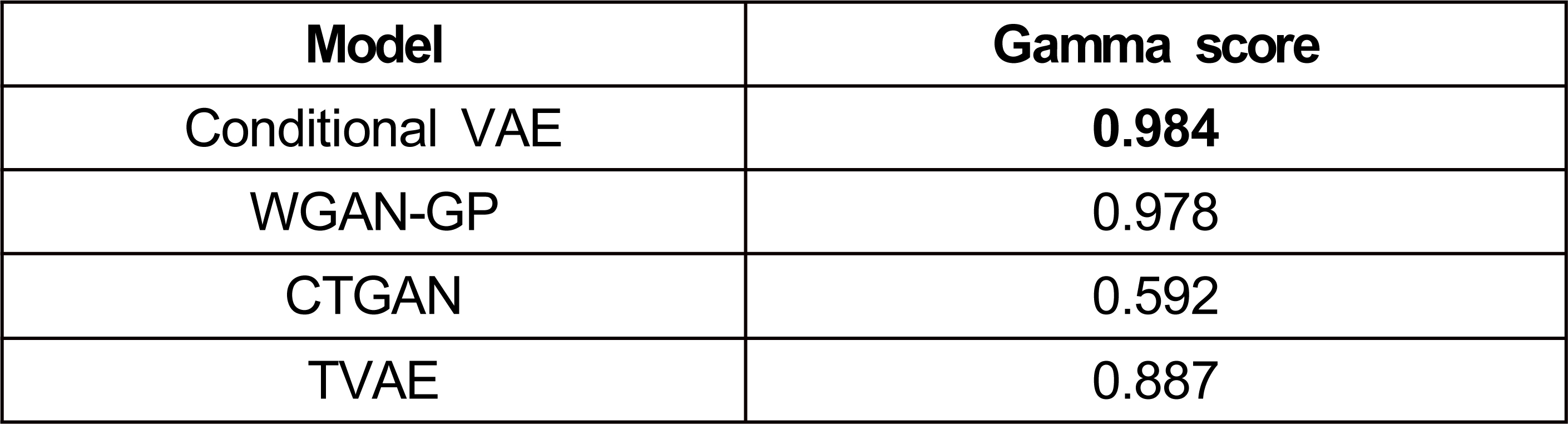
표 2는 각각의 모델이 생성한 합성 유전자 발현 데이터에 대한 gamma score 계산 결과를 나타낸 테이블이다.
각 모델의 gamma score 계산 결과Table 2. Gamma score calculation results for each model
표 2의 굵은 숫자는 가장 높은 gamma score를 의미한다. 계산 결과 Conditional VAE기반 모델이 0.984로 가장 높은 성능을 보였다. WGAN-GP 기반 모델보다 더 높은 유사도를 보였으며 CTGAN, TVAE와는 큰 차이를 보여 통계적인 지표를 보았을 때 본 논문의 Conditional VAE기반 유전자 발현량 생성 모델이 원래 데이터의 특성을 잘 반영한 합성 데이터를 생성하였음을 알 수 있었다.
2. UMAP
그림 3, 4, 5, 6은 차례대로 Conditional VAE, WGAN- GP, CTGAN, TVAE가 생성한 합성 유전자 발현 데이터를 테스트 데이터셋과 함께 UMAP으로 시각화한 그림이다. 합성 유전자 데이터가 실제 데이터의 특성을 모방하여 잘 만들어졌다면 3가지 특징을 만족해야 한다. 첫째, 조직별로 유전자 발현량 패턴이 차이가 많이 나기 때문에 UMAP 그림상에서 조직별로 클러스터가 생겨야 한다. 둘째, 같은 조직에서 정상샘플과 암샘플의 발현량 패턴 또한 차이가 많이 나기 때문에 정상샘플과 암샘플간의 클러스터가 생겨야 한다. 마지막으로 합성 데이터와 테스트 데이터 간에는 구분이 어려워야 하기 때문에 클러스터가 생기지 않아야 한다. 그림 3,4,5,6은 각각의 특징을 확인하기 위해 순서대로 그려 졌으며 조직별로 잘 구분 되는지, 암/정상 여부를 잘 구분 하는지, 테스트/합성 데이터간의 구분이 어려운지를 나타낸다.

Landmark gene RNA-seq에 대한 Conditional VAE의 합성 유전자 발현 데이터의 UMAP 그림Fig. 3. UMAP representation of synthetic gene expression data trained landmark gene RNA-seq with Conditional VAE

Landmark gene RNA-seq에 대한 WGAN-GP기반 모델의 합성 유전자 발현 데이터의 UMAP 그림Fig. 4. UMAP representation of synthetic gene expression data trained landmark gene RNA-seq with WGAN-GP

Landmark gene RNA-seq에 대한 CTGAN의 합성 유전자 발현 데이터의 UMAP 그림Fig. 5. UMAP representation of synthetic gene expression data trained landmark gene RNA-seq with CTGAN

Landmark gene RNA-seq에 대한 TVAE의 합성 유전자 발현 데이터의 UMAP 그림Fig. 6. UMAP representation of synthetic gene expression data trained landmark gene RNA-seq with TVAE
시각화한 결과 Conditional VAE기반 모델과 WGAN-GP 기반 모델이 CTGAN, TVAE보다 조직별, 암, 정상 여부별로 클러스터를 더 잘 생성하여 생물학적으로 의미 있는 합성 데이터를 생성하는 것을 확인하였다. CTGAN, TVAE는 조직별로 유전자 발현의 특징을 구분하여 학습하는 성능이 상대적으로 떨어졌으며 암, 정상 여부를 구분하는 성능에 대해서도 성능이 떨어지는 것을 확인하였다.
VI. 결 론
본 논문에서는 Conditional VAE을 기반으로 하여 합성 유전자 발현 데이터를 생성할 수 있는 모델을 만들었다. TCGA와 GTEx 데이터셋을 15개의 공통 조직으로 결합한 RNA-seq 데이터셋을 만들고 L1000의 랜드마크 유전자로 학습하여 합성 유전자 발현 데이터의 품질을 비교했다.
결론적으로 본 논문에서 만든 모델이 WGAN-GP에 기반한 이전 연구와 정형 데이터 생성 모델인 CTGAN, TVAE보다 생물학적, 통계학적으로 더 유의미한 유전자 발현 데이터를 생성할 수 있음을 보였다. 정형 데이터 생성에 특화된 모델인 CTGAN, TVAE의 비교에서는 유전자 발현 데이터와 같은 고차원의 정형 데이터에서는 한계가 있고 여전히 합성 유전자 발현 데이터를 in silico로 생성할 수 있는 모델의 연구가 중요함을 확인하였다.
본 논문에서 진행한 연구를 기반으로 생물 의료학적 사전 지식을 활용하여 Conditional VAE의 구조를 바꿔 더 생물학적으로 정교한 합성 데이터를 만드는 후속 연구가 가능하고 결국 합성한 데이터를 활용하여 분석 성능을 높일 수 있는 데이터 증강 방법으로써 사용이 가능한지 적용해 보는 다양한 후속 연구가 가능할 것이라 기대한다.
Notes
References
- Lee, Su-min, Recent Development of Next Generation Sequence Analysis (NGS) Technology and Future Research Direction, (2014), BRIC VIEW, 2014-T05.
-
The Cancer Genome Atlas Research Network, et al, Weinstein, J.N., et al , The Cancer Genome Atlas Pan-Cancer analysis project, Nat Genet, (2013), 45, p1113-1120.
[https://doi.org/10.1038/ng.2764]
-
Aguet, F., et al , The GTEx consortium atlas of genetic regulatory effects across human tissues, Science, (2019), 369, p1318-1330.
[https://doi.org/10.1126/science.aaz1776]
-
Rhee, Je-Keun, Prediction for Periodontal Disease using Gene Expression Profile Data based on Machine Learning, Journal of the Korea Institute of Information and Communication Engineering, (2019), 23(8), p903-909.
[https://doi.org/10.6109/jkiice.2019.23.8.903]
-
Li, Y., Umbach, D.M., Krahn, J.M., et al , Predicting tumor response to drugs based on gene-expression biomarkers of sensitivity learned from cancer cell lines, BMC Genomics, (2021), 22, No.272.
[https://doi.org/10.1186/s12864-021-07581-7]
-
Wang, L., Oh, W., Zhu, J, Disease-specific classification using deconvoluted whole blood gene expression, Sci Rep, (2016), 6, No.32976.
[https://doi.org/10.1038/srep32976]
-
Jin, Ting, Nguyen, Nam D, Talos, Flaminia, Wang, Daifeng, ECMarker: interpretable machine learning model identifies gene expression biomarkers predicting clinical outcomes and reveals molecular mechanisms of human disease in early stages, Bioinformatics, (2021, April, 15), 37(8), p1115-1124.
[https://doi.org/10.1093/bioinformatics/btaa935]
-
Kingma, DP, Welling, M, Auto-encoding variational bayes, ICLR, (2014).
[https://doi.org/10.48550/arXiv.1312.6114]
-
Jia, P., Hu, R., Pei, G., et al , Deep generative neural network for accurate drug response imputation, Nat Commun, (2021), 12, No.1740.
[https://doi.org/10.1038/s41467-021-21997-5]
-
Gulrajani, I., Ahmed, F., Arjovsky, M., Dumoulin, V., Courville, A, Improved training of wasserstein gans, NIPS, (2017), 30.
[https://doi.org/10.48550/arXiv.1704.00028]
-
Viñas, R., Andrés-Terré, H., Liò, P., Bryson, K., Adversarial generation of gene expression data, Bioinformatics, (2022, February), 38(3), p730-737.
[https://doi.org/10.1093/bioinformatics/btab035]
-
Kingma, D.P, Rezende, D.J, Mohamed, S, Welling, M, Semi-supervised learning with deep generative models, NIPS, (2014), 27.
[https://doi.org/10.48550/arXiv.1406.5298]
-
Xu, L., Skoularidou, M., Cuesta-Infante, A., Veeramachaneni, K, Modeling tabular data using conditional gan, NIPS, (2019), 32.
[https://doi.org/10.48550/arXiv.1907.00503]
-
Goodfellow, I.J., Pouget-Abadie, J., Mirza, M., Xu, B., Warde-Farley, D., Ozair, S., Courville, A.C., Bengio, Y., Generative adversarial nets, Advances in Neural Information Processing Systems, (2014), p2672-2680.
[https://doi.org/10.48550/arXiv.1406.2661]
-
Arjovsky, M., Chintala, S., Bottou, L., Wasserstein Generative Adversarial Networks, Proceedings of the 34th International Conference on Machine Learning, (2017), p214-223.
[https://doi.org/10.48550/arXiv.1701.07875]
-
Weaver, N., Lipschitz algebras, World Scientific, (2018).
[https://doi.org/10.1142/4100]
- Mirza, M., Osindero, S., Conditional Generative Adversarial Nets, (2014), ArXivPrePrint ArXiv:1411.1784.
-
Chen, Yifei, Li, Yi, Narayan, Rajiv, Subramanian, Aravind, Xie, Xiaohui, Gene expression inference with deep learning, Bioinformatics, (2016, June), 32(12), p1832-1839.
[https://doi.org/10.1093/bioinformatics/btw074]
-
Wang, Q., et al , Unifying cancer and normal RNA sequencing data from different sources, Sci Data, (2018), 5, No.180061.
[https://doi.org/10.1038/sdata.2018.61]
- McInnes, L., et al , UMAP: Uniform Manifold Approximation and Projection for dimension reduction, (2018), ArXivPrePrint ArXiv:1802.03426.

봉 현 수
- 2020년 ~ 현재 : 명지대학교 데이터테크놀로지학과 학사과정
- 주관심분야 : 딥러닝, 생물정보학, 유전체학

오 민 식
- 2015년 : 서울대학교 컴퓨터공학부 학사
- 2021년 : 서울대학교 컴퓨터공학부 박사
- 2021 ~ 2022년 : 서울대학교 지능형 컴퓨팅 사업단 연수연구원
- 2022년 ~ 현재 : 명지대학교 융합소프트웨어학부 데이터테크놀로지전공 조교수
- 주관심분야 : 생물정보학, 딥러닝, 머신러닝